Maximum likelihood by hand
Klaus Schliep, Iris Bardel-Kahr
Graz University of Technology, University of Grazklaus.schliep@gmail.com
2026-05-19
Source:vignettes/MLbyHand.Rmd
MLbyHand.RmdMaximum likelihood by hand
With the function pml_bb from phangorn (Schliep 2011) a lot of steps have become easier
and shorter. If you want to have more control over all of the used
parameters, it is also possible to use the older functions,
e.g. optim_pml. The data is the same as in the vignette
Estimating phylogenetic trees with phangorn:
library(ape)
library(phangorn)
fdir <- system.file("extdata/trees", package = "phangorn")
primates <- read.phyDat(file.path(fdir, "primates.dna"),
format = "interleaved")As a starting tree, we calculate a neighbor joining tree:
fit <- pml(treeNJ, data=primates)
fit## model: JC
## loglikelihood: -3075
## unconstrained loglikelihood: -1230
## Total tree length: 4.056
## (expected number of substituions per site)
## Minimal tree length: 3.216
## (observed substitutions per site)
##
## Rates:
## a <-> c : 1
## a <-> g : 1
## a <-> t : 1
## c <-> g : 1
## c <-> t : 1
## g <-> t : 1
##
## Base frequencies:
## a c g t
## 0.25 0.25 0.25 0.25The function pml returns an object of class
pml. This object contains the data, the tree and many
different parameters of the model like the likelihood. There are many
generic functions for the class pml available, which allow
the handling of these objects.
methods(class="pml")## [1] AICc anova BIC glance logLik plot print simSeq
## [9] terraces update vcov
## see '?methods' for accessing help and source codeThe object fit just estimated the likelihood for the tree it got
supplied, but the branch length are not optimized for the Jukes-Cantor
(Jukes and Cantor 1969) model yet, which
can be done with the function optim.pml.
## 'log Lik.' -3068 (df=25)With the default values pml will estimate a Jukes-Cantor
model. That means equal base frequencies and all transition rates are
equal. The generic function update allows to change
parameters manually. This is not what we usually want to do. However we
might want to supply a different tree or change the number of rate
categories.
## model: F81+G(4)+I
## loglikelihood: -3037
## unconstrained loglikelihood: -1230
## Total tree length: 4.221
## (expected number of substituions per site)
## Minimal tree length: 3.237
## (observed substitutions per site)
## Proportion of invariant sites: 0.2
## Model of rate heterogeneity: Discrete gamma model
## Number of rate categories: 4
## Shape parameter: 1
## Rate Proportion
## 1 0.0000 0.2
## 2 0.1712 0.2
## 3 0.5959 0.2
## 4 1.2500 0.2
## 5 2.9829 0.2
##
## Rates:
## a <-> c : 1
## a <-> g : 1
## a <-> t : 1
## c <-> g : 1
## c <-> t : 1
## g <-> t : 1
##
## Base frequencies:
## a c g t
## 0.37481 0.40160 0.03911 0.18448In the line above we changed the model to a (discrete) rate across site model with 4 rate categories (using the default shape parameter of 1), to 0.2 invariant sites and supply empirical base frequencies.
fitGTR <- optim.pml(fitF81, model="GTR", optInv=TRUE, optGamma=TRUE,
rearrangement = "NNI")
fitGTR## model: GTR+G(4)+I
## loglikelihood: -2611
## unconstrained loglikelihood: -1230
## Total tree length: 30.6
## (expected number of substituions per site)
## Minimal tree length: 3.237
## (observed substitutions per site)
## Proportion of invariant sites: 0.006978
## Model of rate heterogeneity: Discrete gamma model
## Number of rate categories: 4
## Shape parameter: 3.081
## Rate Proportion
## 1 0.0000 0.006978
## 2 0.3982 0.248256
## 3 0.7411 0.248256
## 4 1.0905 0.248256
## 5 1.7983 0.248256
##
## Rates:
## a <-> c : 0.9533
## a <-> g : 64.01
## a <-> t : 0.8128
## c <-> g : 0.00508
## c <-> t : 24.79
## g <-> t : 1
##
## Base frequencies:
## a c g t
## 0.37481 0.40160 0.03911 0.18448We will change the model to the GTR + + I model and then optimize all the parameters.
With the control parameters the thresholds for the fitting process
can be changed. Here we want just to suppress output during the fitting
process. For larger trees the NNI rearrangements often get stuck in a
local maximum. We added two stochastic algorithms to improve topology
search. The first (set rearrangement="stochastic") performs
stochastic rearrangements similar as in (Nguyen
et al. 2015), which makes random NNI permutation to the tree,
which than gets optimized to escape local optima. The second option
(rearrangement="ratchet") perform the likelihood ratchet
(Vos 2003).
While these algorithms may find better trees they will also take more time.
fitGTR <- optim.pml(fitGTR, model="GTR", optInv=TRUE, optGamma=TRUE,
rearrangement = "stochastic")
fitGTR## model: GTR+G(4)+I
## loglikelihood: -2608
## unconstrained loglikelihood: -1230
## Total tree length: 41.74
## (expected number of substituions per site)
## Minimal tree length: 3.315
## (observed substitutions per site)
## Proportion of invariant sites: 0.007369
## Model of rate heterogeneity: Discrete gamma model
## Number of rate categories: 4
## Shape parameter: 2.994
## Rate Proportion
## 1 0.0000 0.007369
## 2 0.3917 0.248158
## 3 0.7366 0.248158
## 4 1.0905 0.248158
## 5 1.8109 0.248158
##
## Rates:
## a <-> c : 0.5937
## a <-> g : 61.01
## a <-> t : 0.4768
## c <-> g : 0.004348
## c <-> t : 21.35
## g <-> t : 1
##
## Base frequencies:
## a c g t
## 0.37481 0.40160 0.03911 0.18448Model comparison
We can compare nested models for the JC and GTR + + I model using likelihood ratio statistic
anova(fitJC, fitGTR)## Likelihood Ratio Test Table
## Log lik. Df Df change Diff log lik. Pr(>|Chi|)
## 1 -3068 25
## 2 -2608 35 10 921 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1with the Shimodaira-Hasegawa test
SH.test(fitGTR, fitJC)## Trees ln L Diff ln L p-value
## [1,] 1 -2608 0.0 0.5043
## [2,] 2 -3068 460.5 0.0000or with the AIC
AIC(fitJC)## [1] 6187
AIC(fitGTR)## [1] 5286
AICc(fitGTR)## [1] 5298
BIC(fitGTR)## [1] 5406Bootstrap
At last we may want to apply standard bootstrap to test how well the edges of the tree are supported. This has already been shown in the vignette Estimating phylogenetic trees with phangorn.
bs <- bootstrap.pml(fitJC, bs=100, optNni=TRUE)Now we can plot the tree with the bootstrap support values on the
edges and also look at consensusNet to identify potential
conflict.
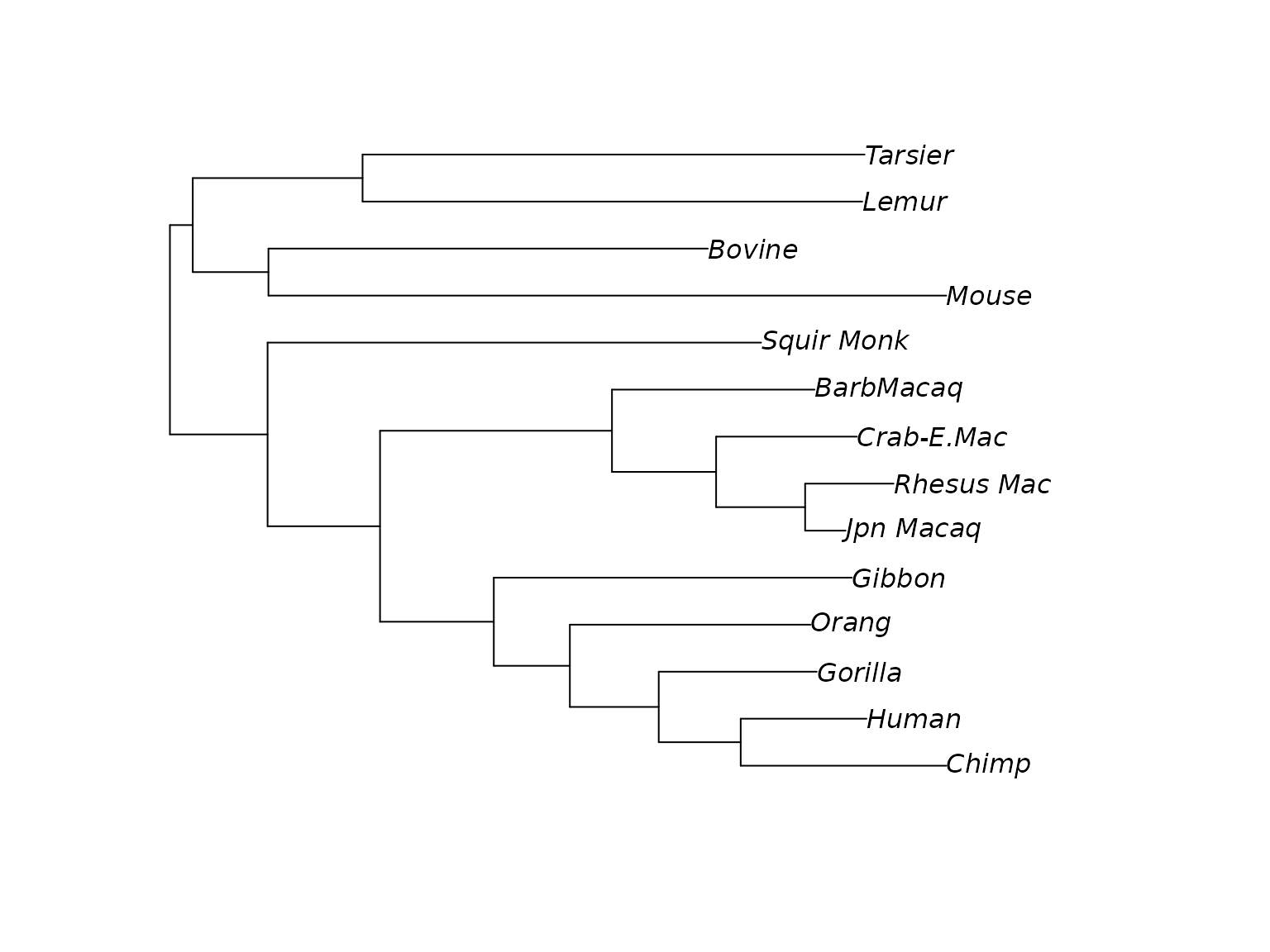
Tree with bootstrap support. Unrooted tree (midpoint rooted) with bootstrap support values.
cnet <- consensusNet(bs, p=0.2)
plot(cnet, show.edge.label=TRUE)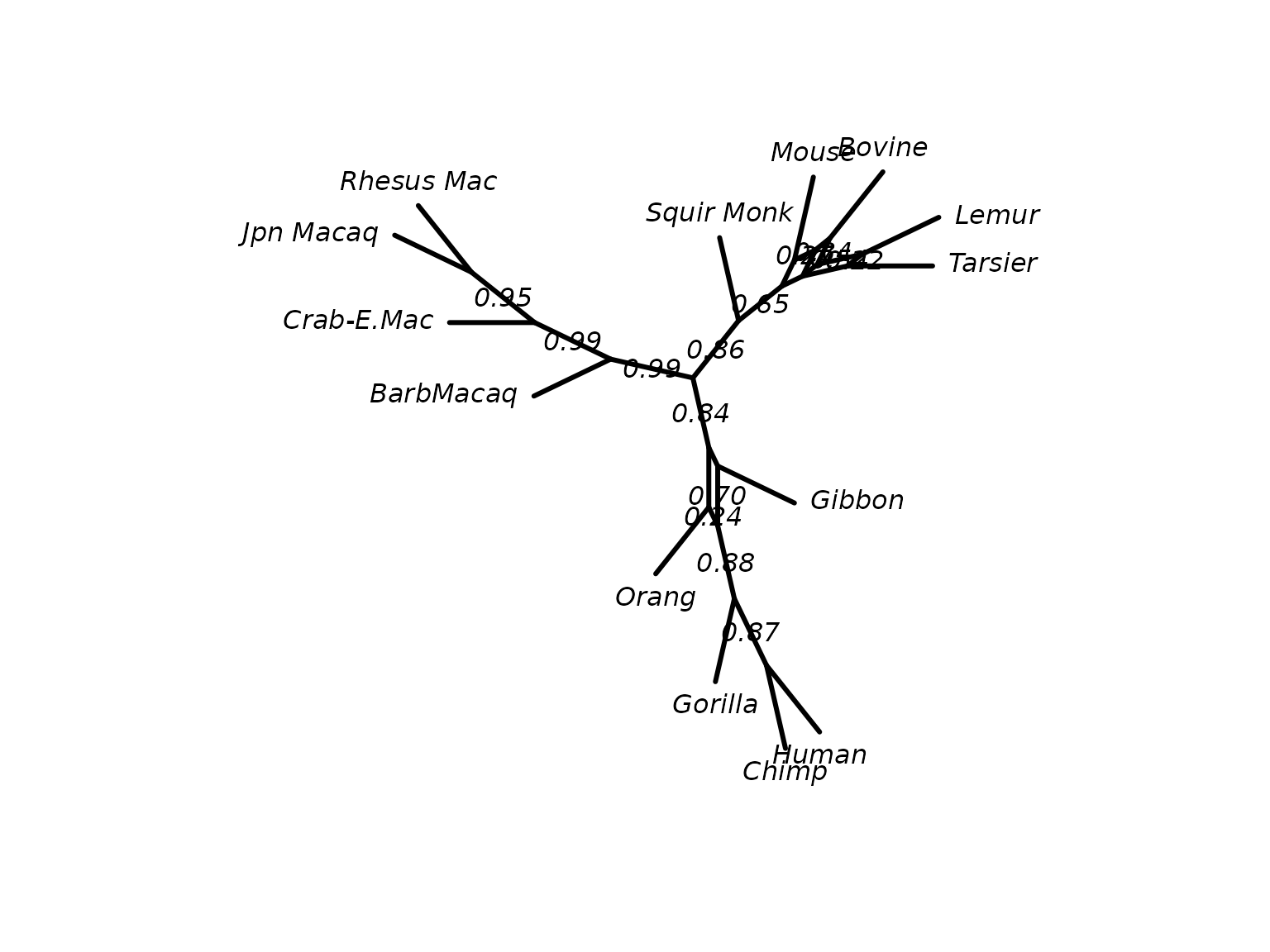
ConsensusNet from the bootstrap sample.
Generating trees
phangorn has several functions to generate tree topologies,
which may are interesting for simulation studies. allTrees
computes all possible bifurcating tree topologies either rooted or
unrooted for up to 10 taxa. One has to keep in mind that the number of
trees is growing exponentially, use howmanytrees from
ape as a reminder.
trees <- allTrees(5)
par(mfrow=c(3,5), mar=rep(0,4))
for(i in 1:15)plot(trees[[i]], cex=1, type="u")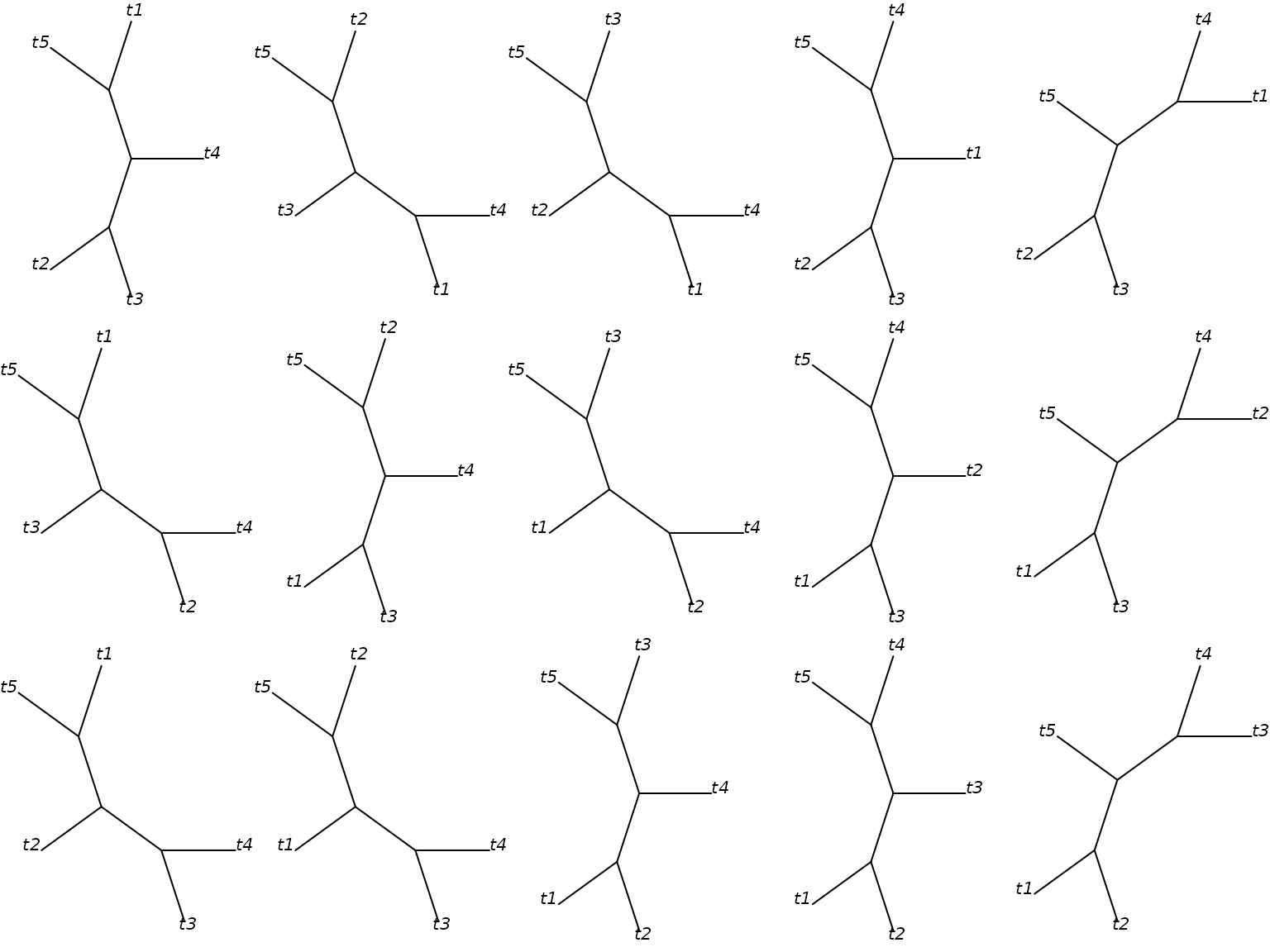
All 15 unrooted trees with five tip labels.
nni returns a list of all trees which are one nearest
neighbor interchange away.
nni(trees[[1]])## 4 phylogenetic treesrNNI and rSPR generate trees which are a
defined number of NNI (nearest neighbor interchange) or SPR (subtree
pruning and regrafting) away.
Session info
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] future_1.70.0 phangorn_2.12.1.4 ape_5.8-1
##
## loaded via a namespace (and not attached):
## [1] Matrix_1.7-5 future.apply_1.20.2 jsonlite_2.0.0
## [4] compiler_4.6.0 Rcpp_1.1.1-1.1 parallel_4.6.0
## [7] jquerylib_0.1.4 globals_0.19.1 systemfonts_1.3.2
## [10] textshaping_1.0.5 yaml_2.3.12 fastmap_1.2.0
## [13] lattice_0.22-9 R6_2.6.1 generics_0.1.4
## [16] igraph_2.3.1 knitr_1.51 htmlwidgets_1.6.4
## [19] backports_1.5.1 checkmate_2.3.4 desc_1.4.3
## [22] bslib_0.11.0 rlang_1.2.0 cachem_1.1.0
## [25] xfun_0.57 fs_2.1.0 sass_0.4.10
## [28] otel_0.2.0 cli_3.6.6 progressr_0.19.0
## [31] pkgdown_2.2.0 magrittr_2.0.5 digest_0.6.39
## [34] grid_4.6.0 lifecycle_1.0.5 nlme_3.1-169
## [37] evaluate_1.0.5 listenv_0.10.1 codetools_0.2-20
## [40] ragg_1.5.2 stats4_4.6.0 parallelly_1.47.0
## [43] rmarkdown_2.31 pkgconfig_2.0.3 tools_4.6.0
## [46] htmltools_0.5.9