Phylogenetic trees from morphological data
Iris Bardel-Kahr, Klaus Schliep
University of Graz, Graz University of Technologyklaus.schliep@gmail.com
2026-05-19
Source:vignettes/Morphological.Rmd
Morphological.RmdIn this vignette, we will show how to work with morphological data in phangorn (Schliep 2011). In most cases the different morphological characters or character states are encoded with the numbers 0:9 (or less, if there are less differences). Morphological data can come in different formats. The most common ones are .csv and .nexus.
Load packages
We start by loading the phangorn package and setting a random seed:
## Loading required package: ape
set.seed(9)Load data
The dataset we’re using contains morphological data for 12 mite
species, with 79 encoded characters (Schäffer et
al. 2010). When reading in the .csv file,
row.names = 1 uses the first column (species) as row names.
To get a phyDat object, we have to convert the dataframe
into a matrix with as.matrix.
fdir <- system.file("extdata", package = "phangorn")
mm <- read.csv(file.path(fdir, "mites.csv"), row.names = 1)
mm_pd <- phyDat(as.matrix(mm), type = "USER", levels = 0:7)The data can then be written into a nexus file:
write.phyDat(mm_pd, file.path(fdir, "mites.nex"), format = "nexus")Reading in a nexus file is even easier than reading in a csv file:
mm_pd <- read.phyDat(file.path(fdir, "mites.nex"), format = "nexus", type = "STANDARD")After reading in the nexus file, we have the states 0:9, but the data only has the states 0:7. Here is one possibility to change the contrast matrix:
contrast <- matrix(data = c(1,0,0,0,0,0,0,0,0,
0,1,0,0,0,0,0,0,0,
0,0,1,0,0,0,0,0,0,
0,0,0,1,0,0,0,0,0,
0,0,0,0,1,0,0,0,0,
0,0,0,0,0,1,0,0,0,
0,0,0,0,0,0,1,0,0,
0,0,0,0,0,0,0,1,0,
0,0,0,0,0,0,0,0,1,
1,1,1,1,1,1,1,1,1),
ncol = 9, byrow = TRUE)
dimnames(contrast) <- list(c(0:7,"-","?"),
c(0:7, "-"))
contrast## 0 1 2 3 4 5 6 7 -
## 0 1 0 0 0 0 0 0 0 0
## 1 0 1 0 0 0 0 0 0 0
## 2 0 0 1 0 0 0 0 0 0
## 3 0 0 0 1 0 0 0 0 0
## 4 0 0 0 0 1 0 0 0 0
## 5 0 0 0 0 0 1 0 0 0
## 6 0 0 0 0 0 0 1 0 0
## 7 0 0 0 0 0 0 0 1 0
## - 0 0 0 0 0 0 0 0 1
## ? 1 1 1 1 1 1 1 1 1
mm_pd <- phyDat(mm_pd, type="USER", contrast=contrast)Now that we have our data, we can start the analyses.
Parsimony
For morphological data, one of the most frequently used approaches to
conduct phylogenetic trees is maximum parsimony (MP).
pratchet (as already described in Estimating
phylogenetic trees with phangorn) implements the parsimony ratchet
(Nixon 1999). To create a starting tree,
we can use the function random.addition:
mm_start <- random.addition(mm_pd)This tree can then be given to pratchet:
mm_tree <- pratchet(mm_pd, start = mm_start, minit = 1000, maxit = 10000,
all = TRUE, trace = 0)
mm_tree## 19 phylogenetic treesWith all=TRUE we get all (in this case 19) trees with
lowest parsimony score in a multiPhylo object. Since we we
did a minimum of 1000 iterations, we already have some edge support. Now
we can assign the edge lengths.
mm_tree <- acctran(mm_tree, mm_pd)Branch and bound
In the case of our mites-dataset with 12 sequences, it’s also
possible to use the branch and bound algorithm (Hendy and Penny 1982) to find all most
parsimonious trees. With bigger datasets it is definitely recommended to
use pratchet.
mm_bab <- bab(mm_pd, trace = 0)
mm_bab## 37 phylogenetic treesRoot trees
If we want our unrooted trees to be rooted, we have the possibility
to use midpoint to perform midpoint rooting. Rooting the
trees with a specific species (we chose C. cymba here) can be
done with the function root from the ape package
(Paradis and Schliep 2019). To save the
correct node labels (edge support), it’s important to set
edgelabel=TRUE.
mm_tree_rooted <- root(mm_tree, outgroup = "C._cymba", resolve.root = TRUE,
edgelabel = TRUE)Plot trees
With plotBS we plot a tree with their respective edge
support. It is also possible to save the plots as .pdf (or
various other formats, e.g. svg, png, tiff) file. digits is
an argument to determine the number of digits shown for the bootstrap
values.
Consensus tree
To look at the consensus tree of our 19 trees from
pratchet, or of our 37 most parsimonious trees from
bab, we can use the consensus function from
ape.
# unrooted pratchet tree
mm_cons <- consensus(mm_tree)
# rooted pratchet tree
mm_cons_root <- consensus(mm_tree_rooted, rooted = TRUE)
# branch and bound, we root the consensus tree in the same step
mm_bab_cons <- root(consensus(mm_bab), outgroup = "C._cymba",
resolve.root = TRUE, edgelabel = TRUE)
plot(mm_cons, main="Unrooted pratchet consensus tree")
plot(mm_cons_root, main="Rooted pratchet consensus tree")
plot(mm_bab_cons, main="Rooted bab consensus tree")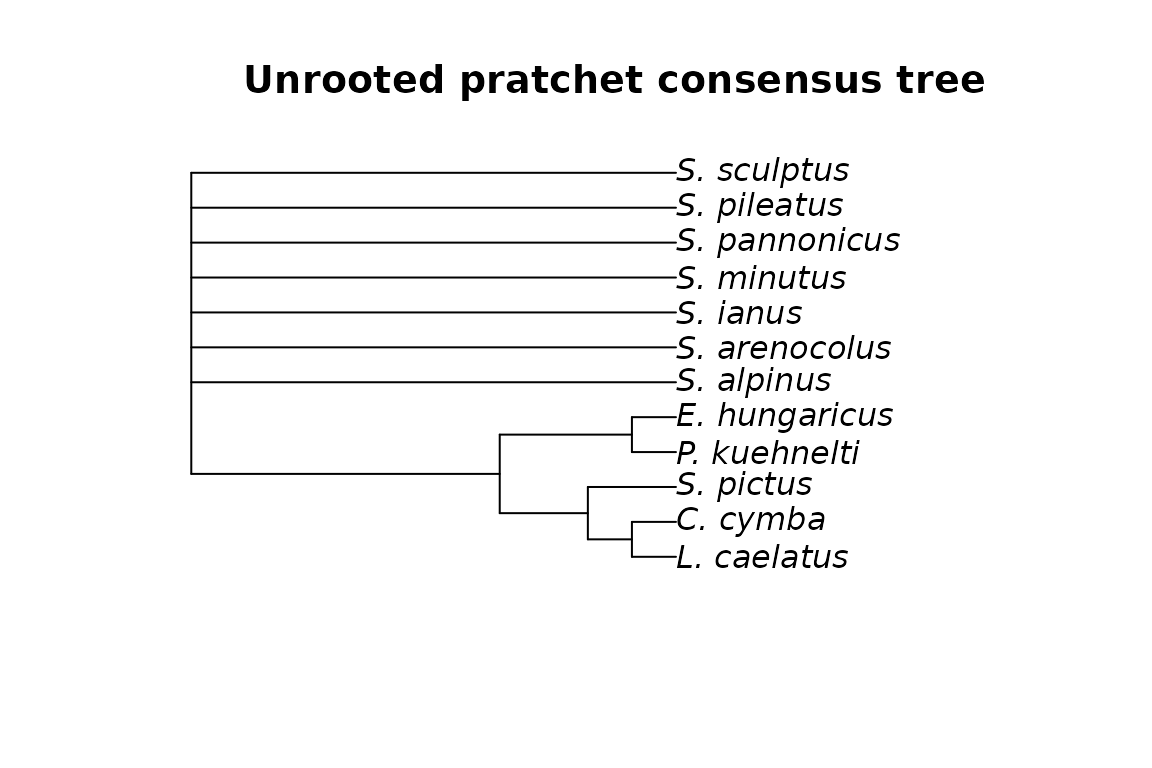
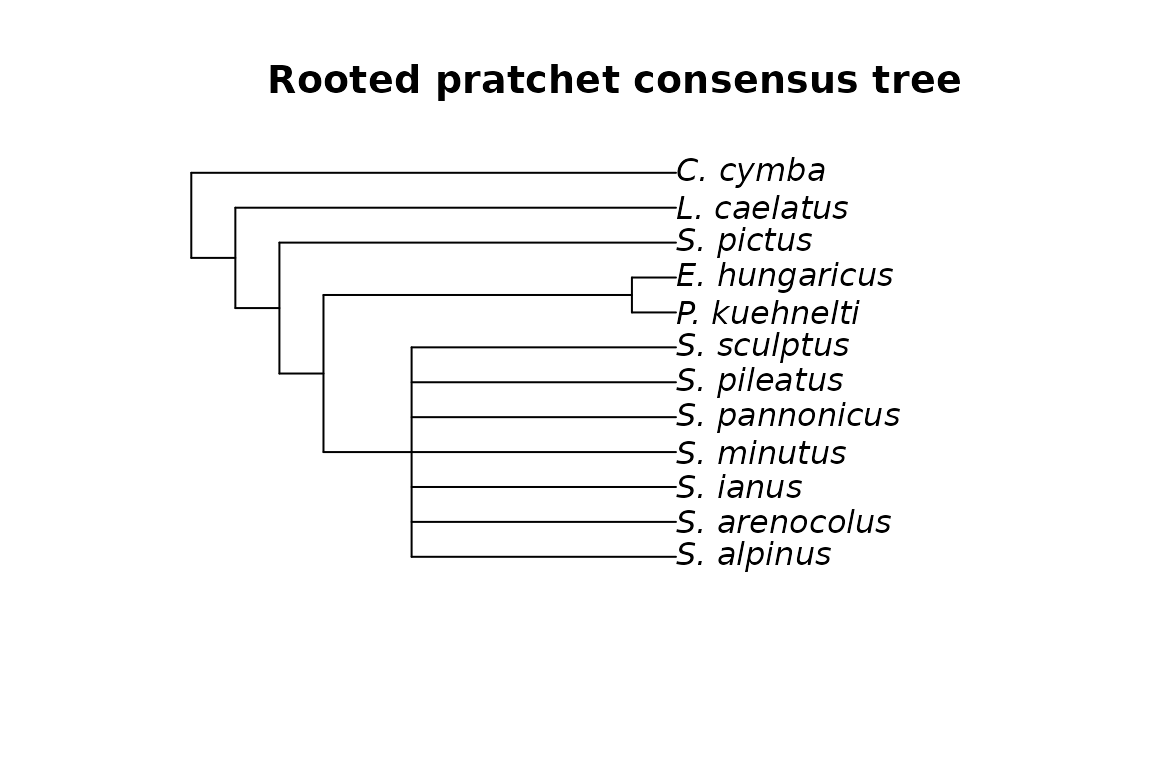
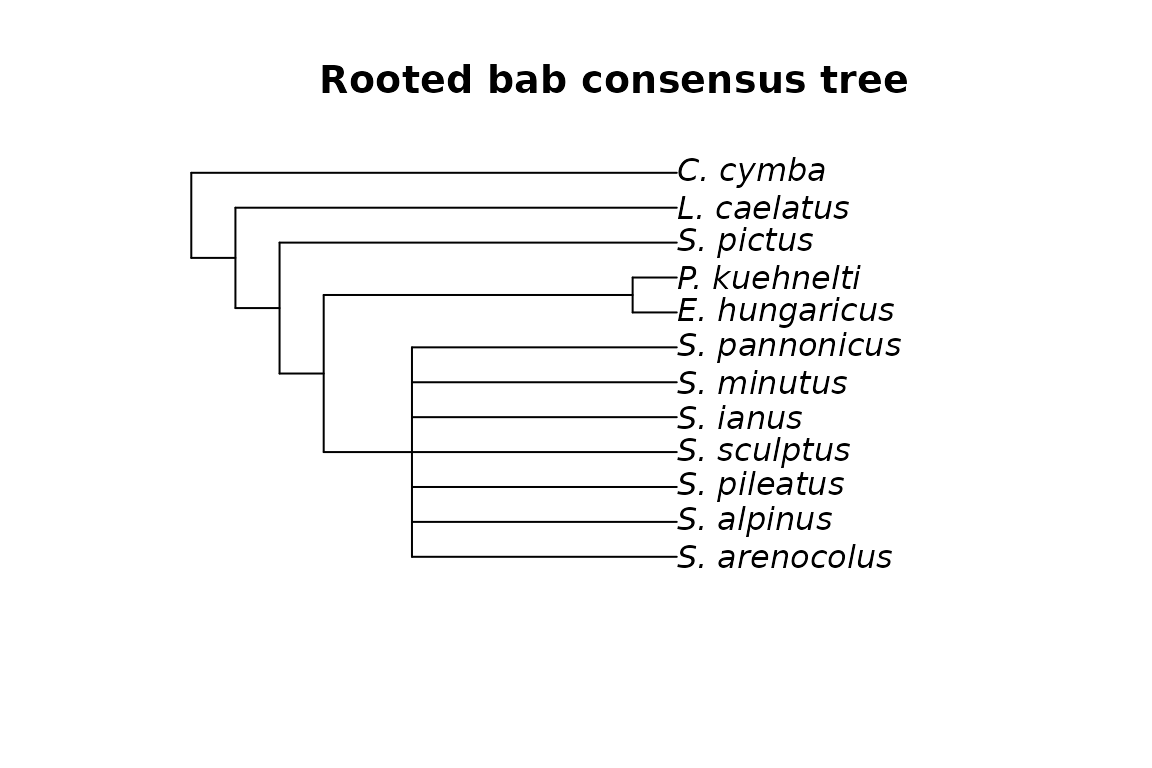
Unrooted and rooted consensus trees of the mites dataset with MP.
We can clearly see that, as expected, the two rooted trees have the same topology.
Session info
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] phangorn_2.12.1.4 ape_5.8-1
##
## loaded via a namespace (and not attached):
## [1] Matrix_1.7-5 future.apply_1.20.2 jsonlite_2.0.0
## [4] compiler_4.6.0 Rcpp_1.1.1-1.1 parallel_4.6.0
## [7] jquerylib_0.1.4 globals_0.19.1 systemfonts_1.3.2
## [10] textshaping_1.0.5 yaml_2.3.12 fastmap_1.2.0
## [13] lattice_0.22-9 R6_2.6.1 generics_0.1.4
## [16] igraph_2.3.1 knitr_1.51 htmlwidgets_1.6.4
## [19] backports_1.5.1 checkmate_2.3.4 future_1.70.0
## [22] desc_1.4.3 bslib_0.11.0 rlang_1.2.0
## [25] cachem_1.1.0 xfun_0.57 fs_2.1.0
## [28] sass_0.4.10 otel_0.2.0 cli_3.6.6
## [31] progressr_0.19.0 pkgdown_2.2.0 magrittr_2.0.5
## [34] digest_0.6.39 grid_4.6.0 lifecycle_1.0.5
## [37] nlme_3.1-169 evaluate_1.0.5 listenv_0.10.1
## [40] codetools_0.2-20 ragg_1.5.2 parallelly_1.47.0
## [43] rmarkdown_2.31 pkgconfig_2.0.3 tools_4.6.0
## [46] htmltools_0.5.9