Matrix for morphological characters and character states for 12 species of mites. See vignette '02_Phylogenetic trees from morphological data' for examples to import morphological data.
References
Schäffer, S., Pfingstl, T., Koblmüller, S., Winkler, K. A., Sturmbauer, C., & Krisper, G. (2010). Phylogenetic analysis of European Scutovertex mites (Acari, Oribatida, Scutoverticidae) reveals paraphyly and cryptic diversity: a molecular genetic and morphological approach. Molecular Phylogenetics and Evolution, 55(2), 677–688.
Examples
data(mites)
mites
#> 12 sequences with 79 character and 53 different site patterns.
#> The states are 0 1 2 3 4 5 6 7
# infer all maximum parsimony trees
trees <- bab(mites)
# For larger data sets you might use pratchet instead bab
# trees <- pratchet(mites, minit=200, trace=0, all=TRUE)
# build consensus tree
ctree <- root(consensus(trees, p=.5), outgroup = "C._cymba",
resolve.root=TRUE, edgelabel=TRUE)
plotBS(ctree, trees)
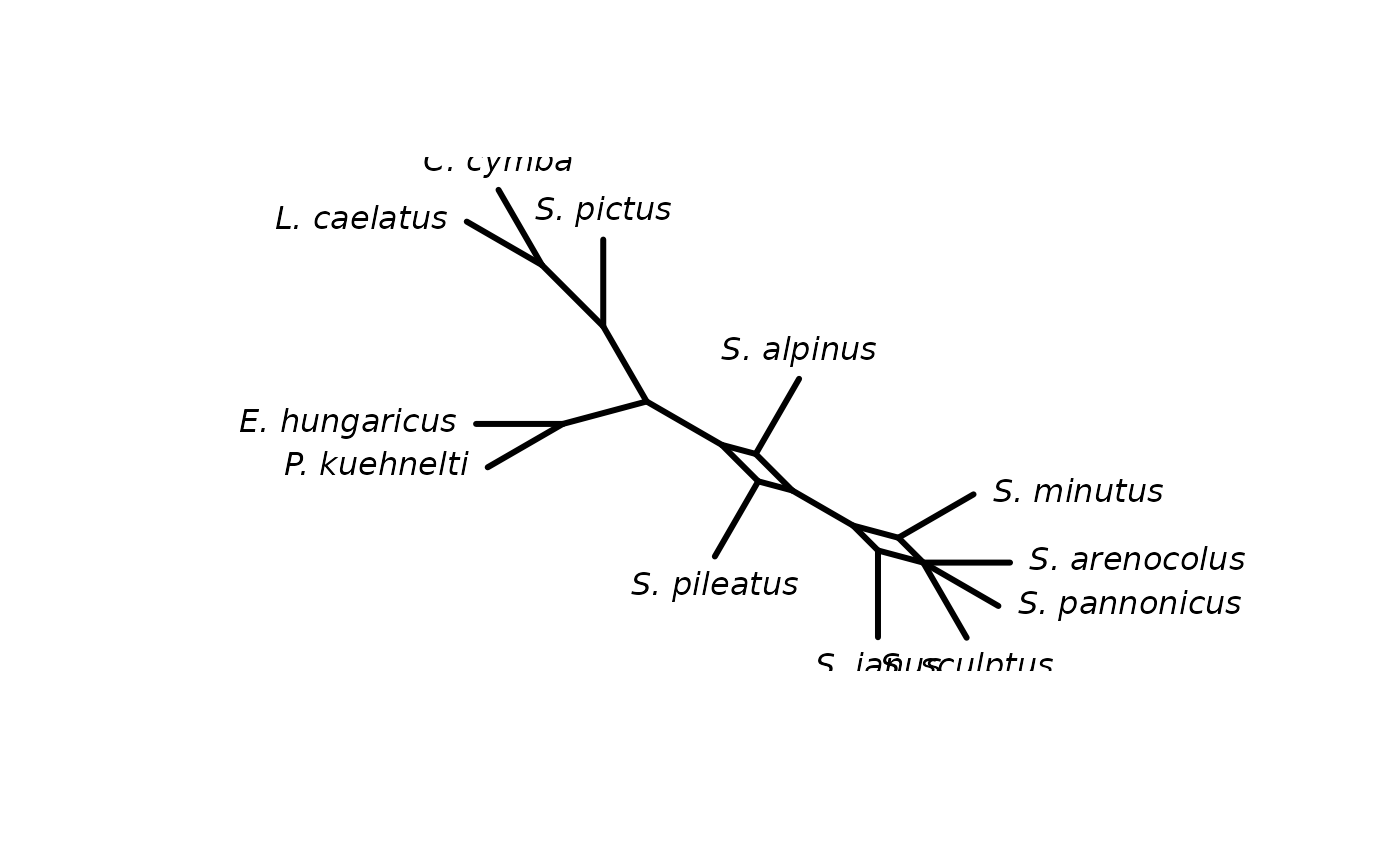
 cnet <- consensusNet(trees)
plot(cnet)
cnet <- consensusNet(trees)
plot(cnet)